/home/lcreteig/miniconda3/envs/SLE/lib/python3.7/site-packages/sklearn/utils/deprecation.py:143: FutureWarning: The sklearn.linear_model.base module is deprecated in version 0.22 and will be removed in version 0.24. The corresponding classes / functions should instead be imported from sklearn.linear_model. Anything that cannot be imported from sklearn.linear_model is now part of the private API.
warnings.warn(message, FutureWarning)
Running the code without the original data
If you want to run the code but don’t have access to the data, run the following instead to generate some synthetic data:
X, y = prep_data(data_all, 'SLE', 'BBD', drop_cols = ["Arthritis","Pleurisy","Pericarditis","Nefritis"] + ["dsDNA1"])dsDNA = X.dsDNA2.values.reshape(-1,1) # only dsDNA from chip as a vector
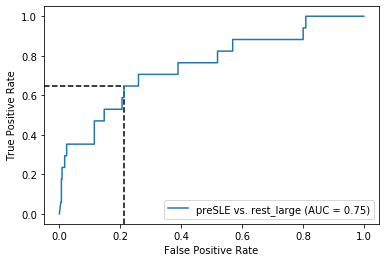
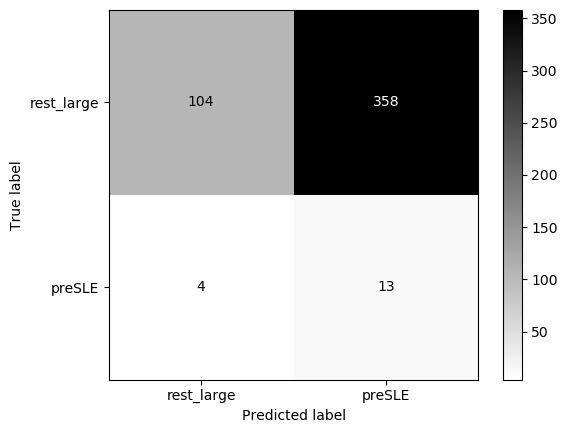
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.98 0.79 0.87 462
preSLE 0.10 0.65 0.17 17
accuracy 0.78 479
macro avg 0.54 0.72 0.52 479
weighted avg 0.95 0.78 0.85 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
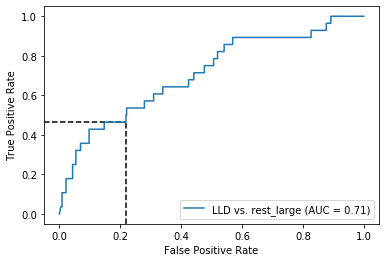
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.96 0.79 0.86 462
LLD 0.12 0.46 0.19 28
accuracy 0.77 490
macro avg 0.54 0.62 0.52 490
weighted avg 0.91 0.77 0.83 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
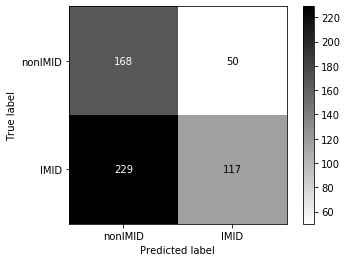
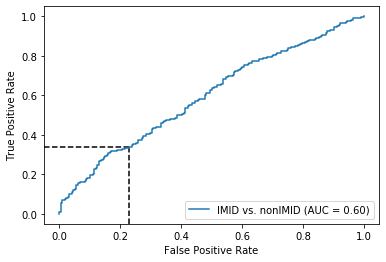
Threshold for classification: 0.5
precision recall f1-score support
nonIMID 0.42 0.77 0.55 218
IMID 0.70 0.34 0.46 346
accuracy 0.51 564
macro avg 0.56 0.55 0.50 564
weighted avg 0.59 0.51 0.49 564
N.B.: "recall" = sensitivity for the group in this row (e.g. IMID); specificity for the other group (nonIMID)
N.B.: "precision" = PPV for the group in this row (e.g. IMID); NPV for the other group (nonIMID)
Logistic regression without dsDNA
no_dsDNA = X.drop(columns='dsDNA2')
np.mean(cross_val_score(clf, no_dsDNA, y, cv=cv, scoring ='roc_auc'))
<matplotlib.axes._subplots.AxesSubplot at 0x7fa2d6eb4810>
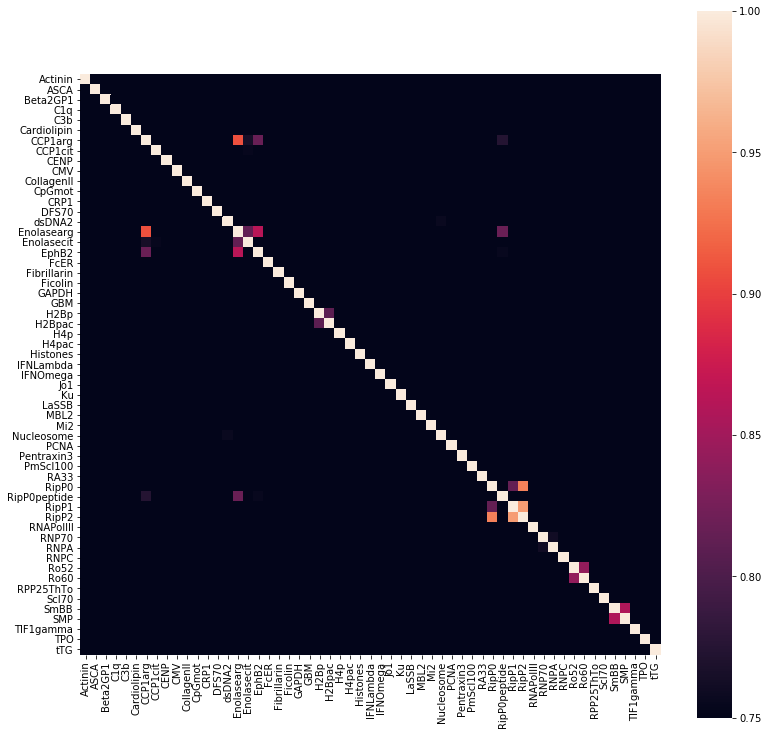
Work down the list of VIFs. Whenever a correlation exceeds threshold, discard feature with highest VIF:
# Set without correlations above .9X_nocor90 = X.drop(columns = ['RipP2', # with RipP1 and RipP0'Enolasearg'# with CCP1arg])
# Set without correlations above .85X_nocor85 = X.drop(columns = ['RipP2', # with RipP1 and RipP0'Enolasearg', # with CCP1arg, EphB2'SmBB'# with SMP])
# Set without correlations above .8X_nocor80 = X.drop(columns = ['RipP2', 'RipP1', # with RipP0'Enolasearg', # with CCP1arg, EphB2, Enolasecit, RipP0peptide'CCP1arg', # with EphB2'SmBB', # with SMP'Ro60', # with Ro52'H2Bp'# with H2Bpac])
Without scaling:
np.mean(cross_val_score(clf, X, y, cv=cv, scoring ='roc_auc'))
0.9111973993393901
Repeat with statsmodels, to get coefs / odds ratios and CIs:
logreg = sm.Logit(y,sm.add_constant(X)).fit()
Optimization terminated successfully.
Current function value: 0.234249
Iterations 13
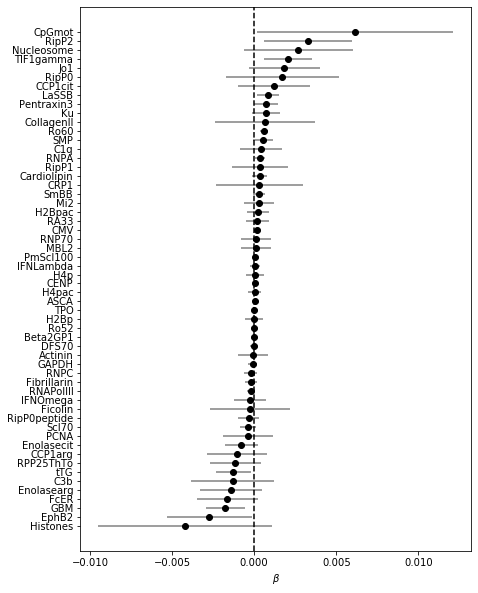
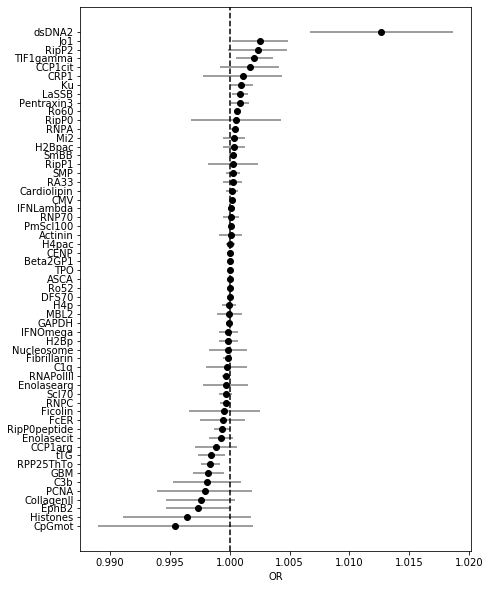
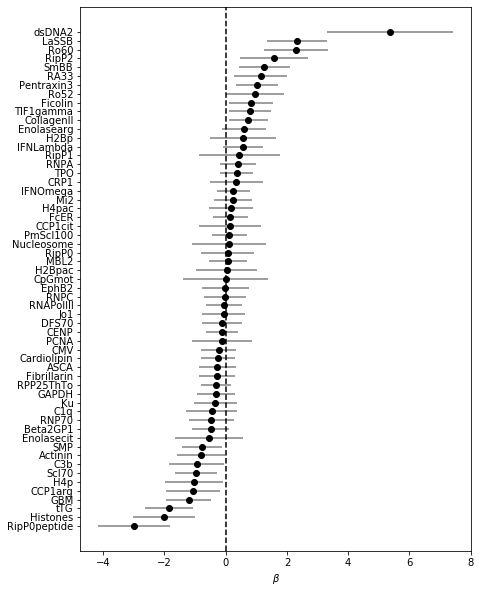
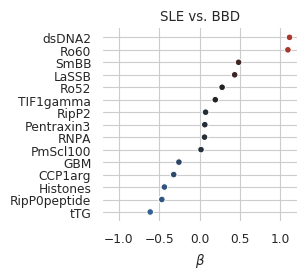
coef_plot(coefs_trf, OR=False) # plotting ORs here is not feasible as dsDNA is so large
Note that once normalized in this way, coefficient for CpGmot becomes very small!
Penalized logistic regression
trf = PowerTransformer(method='box-cox')
Ridge
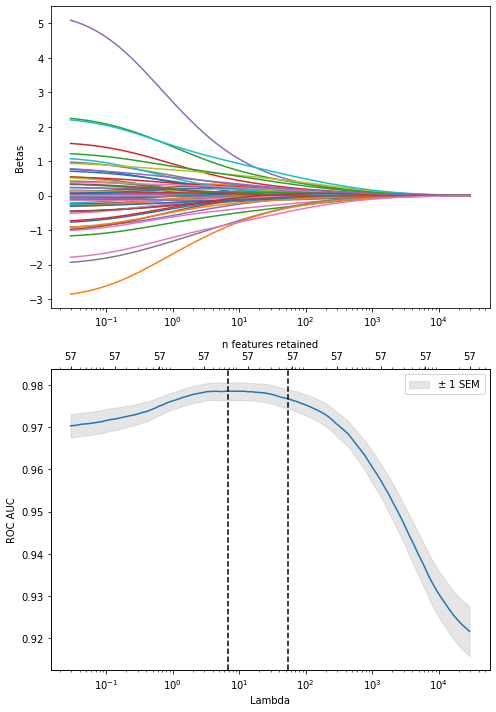
Choose range for regularization:
# for ridge, pick alpha close to zero (if zero, max lambda is infinite. pick alpha=1e-3 to get same as glmnet) # and widen the range between lambda max and min (epsilon)lambda_min, lambda_max = regularization_range(Xp1,y,trf,alpha=1e-2, epsilon=1e-6)
Choose a final value for the regularization parameter with the 1 SE rule (as just choosing the best lambda tends to under-regularize, https://stats.stackexchange.com/questions/138569)
/home/lcreteig/miniconda3/envs/SLE/lib/python3.7/site-packages/sklearn/svm/_base.py:977: ConvergenceWarning: Liblinear failed to converge, increase the number of iterations.
"the number of iterations.", ConvergenceWarning)
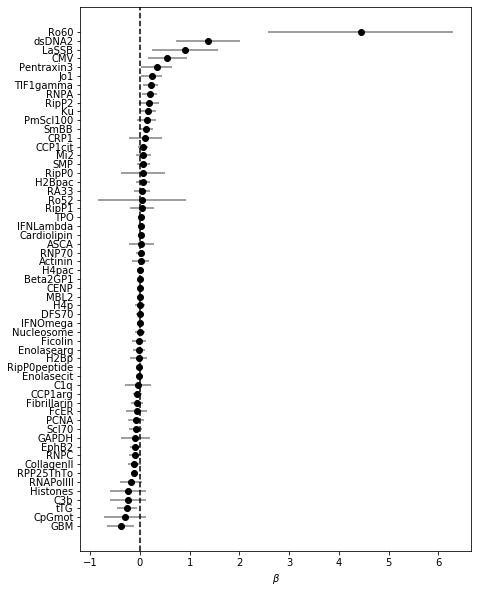
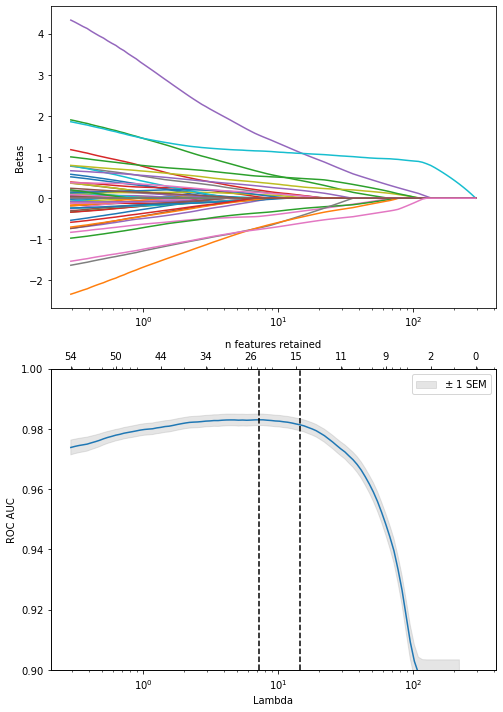
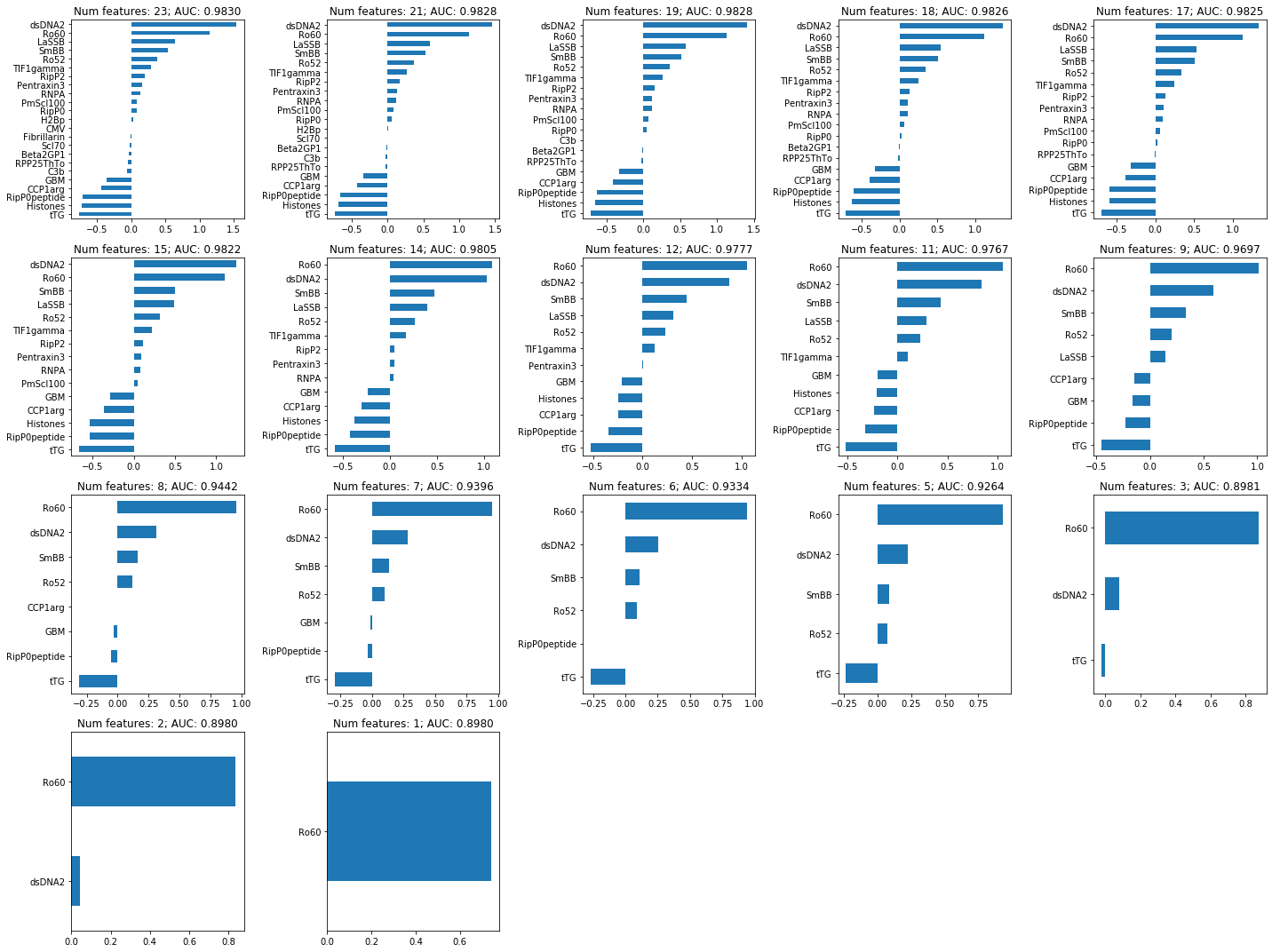
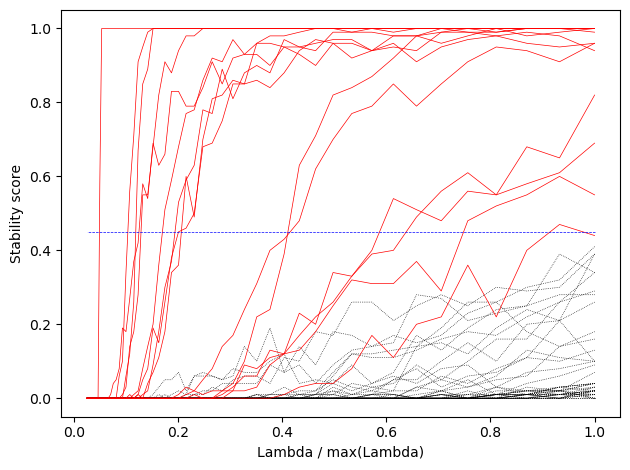
Plot the coefficients for models with different levels of regularization (= number of features retained). Start with the model with the best CV ROC AUC, and work our way down to include less and less features:
If we pick a certain lambda value, the features that get included might vary with a different dataset. We can estimate that variability by taking bootstrap samples of the dataset. Then we can select the features that are included most often
%%timeselector = StabilitySelection(base_estimator=pipe, lambda_name='clf__C', lambda_grid=Cs_lasso[np.argmax(search_lasso.cv_results_["mean_test_score"]):], #range from highest scoring lambda to lambda_max random_state=40) selector.fit(Xp1, y)
CPU times: user 7min 37s, sys: 0 ns, total: 7min 37s
Wall time: 7min 37s
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.96 0.23 0.36 462
preSLE 0.04 0.76 0.07 17
accuracy 0.24 479
macro avg 0.50 0.49 0.22 479
weighted avg 0.93 0.24 0.35 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
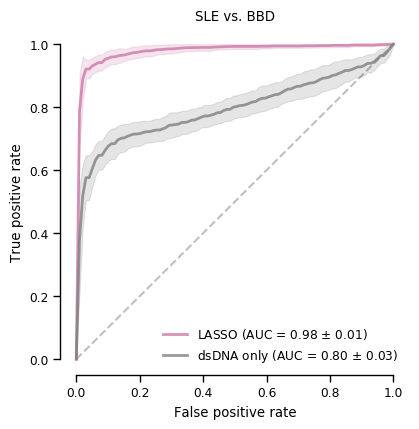
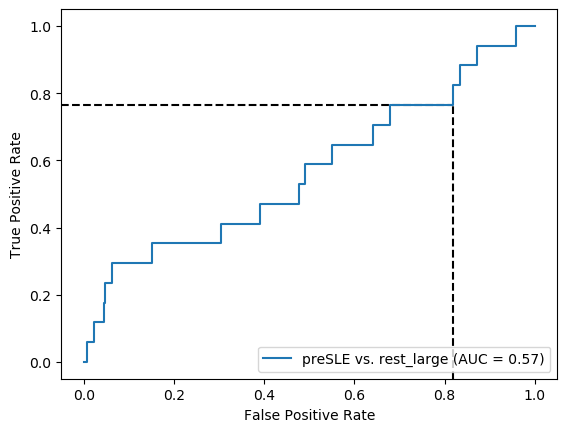
Sensitivity is a bit better than when only using dsDNA, but everything else is worse
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.95 0.23 0.36 462
LLD 0.06 0.82 0.11 28
accuracy 0.26 490
macro avg 0.51 0.52 0.24 490
weighted avg 0.90 0.26 0.35 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
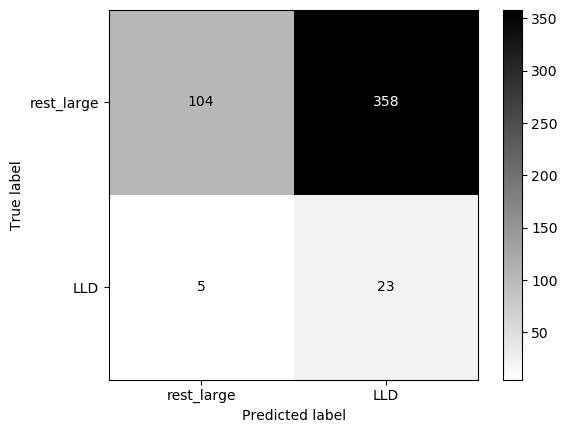
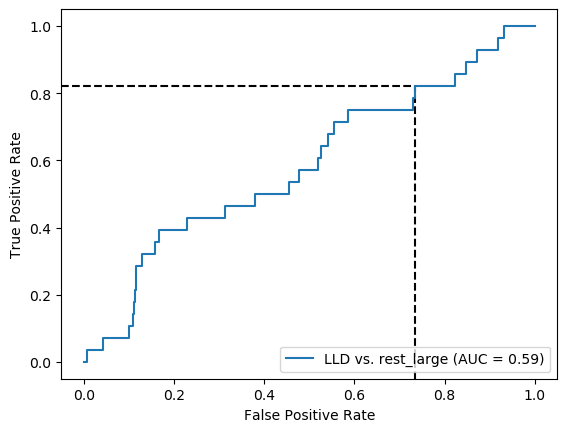
Similar to preSLE. Seems that when trained on blood bank controls, model classifies everyone as a patient
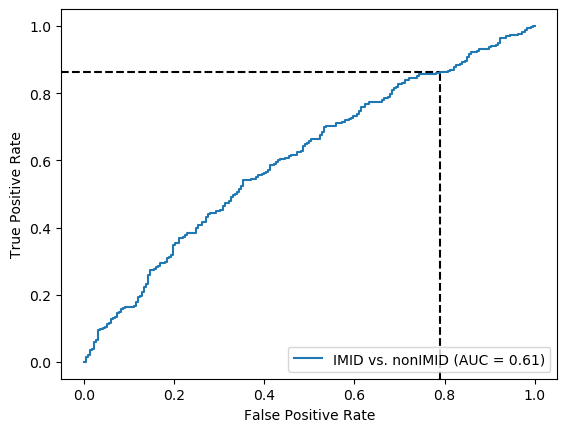
Threshold for classification: 0.5
precision recall f1-score support
nonIMID 0.49 0.21 0.29 218
IMID 0.63 0.86 0.73 346
accuracy 0.61 564
macro avg 0.56 0.54 0.51 564
weighted avg 0.58 0.61 0.56 564
N.B.: "recall" = sensitivity for the group in this row (e.g. IMID); specificity for the other group (nonIMID)
N.B.: "precision" = PPV for the group in this row (e.g. IMID); NPV for the other group (nonIMID)
So the best model tends to favor L1 more than L2, and has a similar lambda value compared to pure L1/L2
search_enet.best_score_
0.9833992316750093
AUC is a bit higher, but similar to LASSO
# Get coefficients for each C/l1_ratiocoefs_enet = np.empty([len(Cs), len(l1l2), len(X.columns)])for i,c inenumerate(Cs):for j,l inenumerate(l1l2): clf_enet.set_params(C=c, l1_ratio = l, warm_start=True) clf_enet.fit(X_trf,y) coefs_enet[i,j,:] = clf_enet.coef_
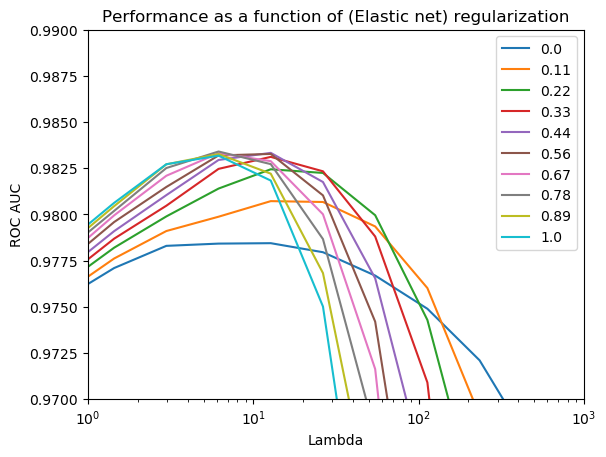
# Plot AUC and n_features for different lambdasscores = search_enet.cv_results_["mean_test_score"].reshape(len(Cs),len(l1l2))lines = plt.plot(1/Cs, scores)plt.legend(lines, np.round(l1l2,2))plt.xscale('log')plt.xlim(1e0, 1e3)plt.ylim([0.97, .99])plt.xlabel('Lambda')plt.ylabel('ROC AUC')plt.title('Performance as a function of (Elastic net) regularization')
Text(0.5, 1.0, 'Performance as a function of (Elastic net) regularization')
Here 0 = Ridge; 1 = LASSO. Max performance is very similar to pure LASSO, but with stronger regularization the hybrid models start to do a little better than pure Ridge or LASSO (but this is a small difference; note the scaling!)
clf_rf = RandomForestClassifier(random_state=40)np.mean(cross_val_score(clf_rf, X, y, cv=cv, scoring ='roc_auc'))
0.9912558838826646
Completely untuned random forest with no feature preprocessing does even better still, but the increase in AUC is not worth the decrease in interpretability… Note though that this performs even a bit better than the (extensively tuned) XGBoost!